
The gene mutations driving cancer have been tracked for the first time
The gene mutations driving cancer have been tracked for the first time in patients back to a distinct set of cells at the root of cancer -- cancer stem cells. The international research team studied a group of patients with myelodysplastic syndromes -- a malignant blood condition which frequently develops into acute myeloid leukemia. The researchers say their findings offer conclusive evidence for the existence of cancer stem cells.
The international research team, led by scientists at the University of Oxford and the Karolinska Institutet in Sweden, studied a group of patients with myelodysplastic syndromes -- a malignant blood condition which frequently develops into acute myeloid leukemia. The researchers say their findings, reported in the journal Cancer Cell, offer conclusive evidence for the existence of cancer stem cells.
HIGHLIGHTS
•
MDS stem cells and progenitors are distinct and hierarchically related
•
Mutations in low-risk MDS originate exclusively in distinct and rare MDS stem cells
•
Mutations preceding AML transformation might confer self-renewal to MDS progenitors
•
del(5q) precedes acquisition of recurrent driver mutations in isolated del(5q) MDS
SUMMARY
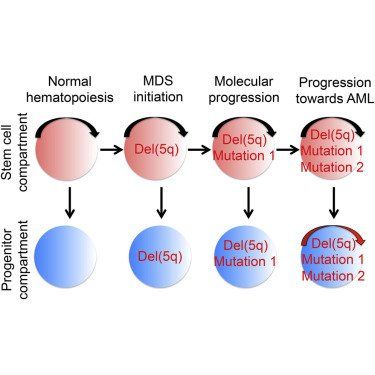
Evidence for distinct human cancer stem cells (CSCs) remains contentious and the degree to which different cancer cells contribute to propagating malignancies in patients remains unexplored. In low- to intermediate-risk myelodysplastic syndromes (MDS), we establish the existence of rare multipotent MDS stem cells (MDS-SCs), and their hierarchical relationship to lineage-restricted MDS progenitors. All identified somatically acquired genetic lesions were backtracked to distinct MDS-SCs, establishing their distinct MDS-propagating function in vivo. In isolated del(5q)-MDS, acquisition of del(5q) preceded diverse recurrent driver mutations. Sequential analysis in del(5q)-MDS revealed genetic evolution in MDS-SCs and MDS-progenitors prior to leukemic transformation. These findings provide definitive evidence for rare human MDS-SCs in vivo, with extensive implications for the targeting of the cells required and sufficient for MDS-propagation.
GRAPHICAL ABSTRACT
SIGNIFICANCE
Experimental evidence supporting the existence of human cancer stem cells (CSCs) remain extensively contested and in vivo fate mapping of candidate human CSCs in patients has not been possible. Through establishment of molecularly and functionally distinct and hierarchically organized stem and progenitor cell compartments in myelodysplastic syndromes (MDS) and backtracking of identified somatic genetic lesions, we establish that rare Lin−CD34+CD38−CD90+CD45RA−MDS cells function as MDS-propagating cells in patients with low- to intermediate-risk MDS. Because their elimination will be essential, and possibly also sufficient, toward eradication of the entire MDS clone, the definitive identification of rare but distinct MDS stem cells will now facilitate development of therapies specifically targeting MDS stem cells.
INTRODUCTION
The concept that human cancers might be propagated exclusively by rare self-renewing cancer stem cells (CSCs), replenishing nontumorigenic cancer cells, has extensive implications for the development of targeted cancer therapies (Clevers, 2011 and Magee et al., 2012). Whereas the existence of cells with human CSC-potential has been supported experimentally in some hematological malignancies (Bonnet and Dick, 1997,Goardon et al., 2011, Jamieson et al., 2004 and Nilsson et al., 2000) and solid tumors (Al-Hajj et al., 2003 and Schatton et al., 2008), the CSC concept has recently been contested in mouse and man (reviewed in Clevers, 2011 and Magee et al., 2012) through multiple studies suggesting that cells with CSC potential, including leukemic stem cells (LSCs), might be neither rare (Kelly et al., 2007 and Quintana et al., 2008) nor phenotypically or molecularly distinct (le Viseur et al., 2008 and Quintana et al., 2008). Of particular relevance, compelling evidence has demonstrated significant, intrinsic limitations of existing human CSC and LSC in vivo assays (Clevers, 2011, Kelly et al., 2007, Magee et al., 2012 and Taussig et al., 2008), failing to reveal the cancer-propagating potential of investigated cancer cell populations as illustrated by a large fraction of patients with acute myeloid leukemia (AML) having no leukemic cells reading out in LSC assays (Pearce et al., 2006).
Regardless, in vitro or in vivo CSC/LSC assays cannot establish to what degree different cancer cells act to propagate the cancer in patients, which ultimately is the most biologically and clinically relevant CSC property. Proposed identities of human CSCs/LSCs therefore await definitive verifications in patients (Clevers, 2011 and Magee et al., 2012), although it will be difficult to apply the genetically engineered lineage-tracing technologies in human malignancies, which recently allowed the definitive identification and fate-mapping of mouse CSCs in vivo (Chen et al., 2012, Driessens et al., 2012 and Schepers et al., 2012).
Myelodysplastic syndromes (MDS) are clonal hematopoietic disorders characterized by inefficient hematopoiesis and frequent progression to AML (Nimer, 2008). Even in low-risk MDS, clonal hematopoiesis already dominates at diagnosis, and clones found in secondary AML originate from the MDS stage of disease (Walter et al., 2012), highlighting the need to specifically target the MDS-initiating clone. Previous studies provided support for del(5q)-MDS originating in hematopoietic stem cells (HSCs; Nilsson et al., 2000), and in vitro and in vivo stem cell (SC) assays have supported that rare CD34+CD38− cells possess MDS-SC potential in low- to intermediate-risk MDS (Nilsson et al., 2000, Nilsson et al., 2002, Pang et al., 2013 and Tehranchi et al., 2010). However, it remains to be established whether CD34+CD38− cells are the only cells with SC potential in MDS because other MDS progenitors, including distinct myeloid progenitor subsets (Manz et al., 2002), have yet to be explored for their MDS-SC potential (Agarwal, 2012, ASH-Workshop, 2010, Nilsson et al., 2000 and Pang et al., 2013). This is particularly relevant because genomic lesions might potentially confer in vivo self-renewal ability to otherwise short-lived myeloid progenitors.
We hypothesized that recently identified recurrent somatic driver-mutations in low- to intermediate-risk MDS (Bejar et al., 2011, Papaemmanuil et al., 2011, Papaemmanuil et al., 2013 and Yoshida et al., 2011) should provide genetic tools to map the identity and fate of MDS-propagating cells in patients in vivo.
RESULTS
Conservation of a Hierarchy of Molecularly and Functionally Distinct Stem and Progenitor Cells in Low- to Intermediate-Risk MDS
Because candidate CSCs/LSCs have frequently not been documented to be molecularly and functionally distinct or hierarchically related to other cancer/leukemic cells (Clevers, 2011, le Viseur et al., 2008 and Magee et al., 2012), a requirement for our fate mapping approach to be informative, we first compared the phenotypic, molecular and functional properties as well as hierarchical relationships of Lineage− (Lin−)CD34+CD38−CD90+CD45RA− candidate MDS-SCs with myeloid-restricted granulocyte-macrophage and megakaryocyte-erythroid progenitors (GMPs and MEPs, respectively; Majeti et al., 2007 and Manz et al., 2002).
In all investigated MDS cases (Table S1 available online), phenotypically defined SCs, GMPs, MEPs, and common myeloid progenitors (CMPs) were identified in agreement with previous studies (Pang et al., 2013 and Will et al., 2012). Each population was preserved at a low frequency despite high clonal involvement (Figures 1A–1D). Absolute numbers of phenotypic SCs and CMPs were expanded and GMPs reduced compared to age-matched controls in isolated del(5q) MDS, other patients with low- to intermediate-risk MDS with del(5q), and in non-del(5q) MDS (Figure 1B). As reported recently for patients with low-risk MDS (Pang et al., 2013), the suppression of GMPs and increase in CMPs became clearer in del(5q) and non-del(5q) MDS cases when assessed relative to the whole Lin−CD34+CD38+ progenitor compartment (Figure 1B). This analysis also revealed that MEPs were relatively suppressed in non-del(5q) cases, but not in cases with del(5q) (Figure 1B). In MDS cases with del(5q), the mean frequency of Lin−CD34+CD38−CD90+CD45RA− SCs with del(5q) was as high as 93.7% and 98.9% as determined by fluorescence in situ hybridization (FISH) and sequencing, respectively (Figures 1C and 1D), suggesting that del(5q) MDS-SCs outcompete normal HSCs. With FISH, del(5q) was found to be high in CMPs, GMPs, MEPs, and SCs, although slightly lower in GMPs and MEPs as determined by sequencing (Figure 1D), suggesting perhaps a slight disadvantage for generation or maintenance of del(5q) GMPs and MEPs.
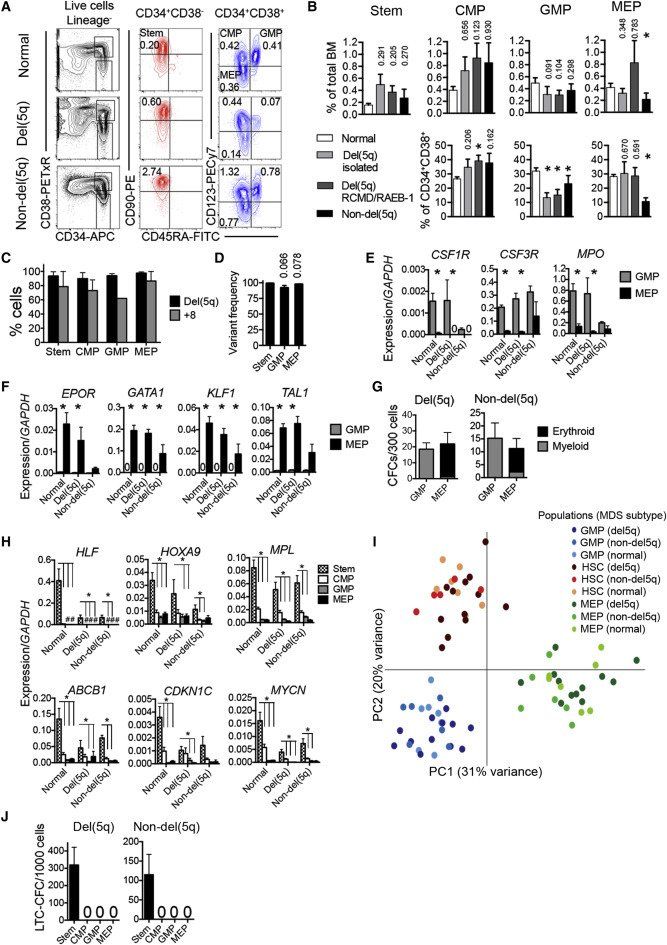
Figure 1.
Conservation of Molecularly and Functionally Distinct Hematopoietic Stem and Progenitor Cells in MDS
(A) FACS profiles of bone marrow stem and progenitor cells in normal age-matched control (top row), and representative del(5q) MDS (patient 1, middle row), and non-del(5q) MDS (patient 68, bottom row) patients. Numbers indicate percentages of total nucleated BM cells.
(B) Mean (SEM) stem/progenitor cell percentages in normal age-matched controls (n = 7) and low- to intermediate-risk isolated del(5q) (n = 9), del(5q) RCMD/RAEB-1 (n = 11) and non-del(5q) (n = 12) MDS in total BM (top row), and Lin−CD34+CD38+ myeloid progenitor compartment (p values against normal controls are shown, ∗p < 0.05).
(C and D) Mean (SEM) del(5q)/trisomy 8 involvement as determined by FISH (C) (del(5q) n = 5, +8 n = 2) and DNA sequencing (D) (n = 7), p values against SCs.
(E and F) Mean (SEM) expression of myeloid (E) and erythroid (F) transcripts within GMPs and MEPs from normal BM (n = 7), del(5q) MDS (n = 6), and non-del(5q) MDS (n = 4). ∗p < 0.05.
(G) Mean (SEM) myeloid and erythroid colony formation of purified GMPs and MEPs from del(5q) MDS (n = 17) and non-del(5q) MDS (n = 7).
(H) Mean (SEM) expression of stem cell transcripts in normal age-matched BM (n = 7), del(5q) MDS (n = 6) and non-del5q MDS (n = 4) stem/progenitor cells. ∗p < 0.05; #, too low to depict.
(I) Principle component analysis (log-transformed) of normal control, del(5q) and non-del(5q) MDS-SCs, GMPs, and MEPs. Each dot represents the indicated cell population from one subject.
(J) Mean (SEM) LTC-CFC formation of purified stem (Lin−CD34+CD38−CD90+CD45RA−) and progenitor cells from del(5q) MDS (n = 15) and non-del(5q) MDS (n = 11).
See also Figure S1 and Table S1.
Because expression of cell surface antigens can be altered in malignant hematopoiesis (le Viseur et al., 2008 and Nguyen et al., 2012), we also interrogated candidate MDS GMPs and MEPs at the molecular and functional level. Candidate MDS GMPs and MEPs displayed transcriptional expression profiles typical of normal GMPs and MEPs (Figures 1E and 1F), and importantly had myeloid- and erythroid-restricted lineage potentials, respectively, in vitro (Figure 1G; Figures S1A and S1B). MDS CMPs (Figure 1A) expressed, as normal CMPs, both lineage programs, which were low to absent in candidate MDS-SCs (Figure S1C). Genes characteristic of normal HSCs were selectively expressed in MDS Lin−CD34+CD38−CD90+CD45RA− SCs (Figure 1H). The distinct molecular signatures of MDS SCs, GMPs, and MEPs was further underpinned by a principle component analysis of RNA sequencing data from purified stem and progenitor cells (Figure 1I), demonstrating that SCs, GMPs, and MEPs from patients with MDS and age-matched normal controls clustered together and distant from the other cell populations.
The ability to sustain long-term generation of MDS myeloid progenitors in vitro (Hogge et al., 1996) was exclusively restricted to Lin−CD34+CD38−CD90+CD45RA− cells and never observed for MDS CMPs, GMPs, or MEPs (Figure 1J; Figure S1D).
Hierarchical Organization of MDS Stem and Progenitor Cells
The distinct molecularly and functionally “lineage-restricted” signatures of MEPs and GMPs in all investigated MDS patients, distinguishing them from the SC-signature of Lin−CD34+CD38−CD90+CD45RA− SCs, implicated a hierarchical relationship between Lin−CD34+CD38−CD90+CD45RA− SCs and myeloid-restricted MEPs and GMPs. We further explored this by investigating the ability of del(5q) Lin−CD34+CD38−CD90+CD45RA− cells to replenish MEPs and GMPs. Because a high fraction of human AMLs fail to reconstitute in xenograft assays (Pearce et al., 2006), it is not surprising that also in most cases of MDS the bone marrow (BM) cells fail to reconstitute immune-deficient mice (Nilsson et al., 2002 and Thanopoulou et al., 2004), although in a recent study SCs from a few monosomy 7 MDS cases did engraft (Pang et al., 2013). In two del(5q) MDS cases, with distinct MEPs, GMPs, and Lin−CD34+CD38−CD90+CD45RA−candidate SCs (Figures 2A–2D; Figures S2A–S2C), multiple mice reconstituted with Lin−CD34+CD38−CD90+CD45RA− cells (Figure 2E; Figure S2D). Not previously investigated (Pang et al., 2013), but of decisive importance for proposing that Lin−CD34+CD38−CD90+CD45RA− MDS cells might be the only MDS-SCs, no reconstitution was observed with purified CMPs, GMPs, MEPs, or CD34neg cells (Figure 2E; Figure S2D). In both patients, Lin−CD34+CD38−CD90+CD45RA− cells only reconstituted myelopoiesis long term, in agreement with B lymphopoiesis being suppressed in MDS (Figure 2F;Figure S2E; Pang et al., 2013 and Sternberg et al., 2005). Reconstituting Lin−CD34+CD38−CD90+CD45RA−SCs sustained a high fraction of CD34+ cells, including clonally involved Lin−CD34+CD38−CD90+CD45RA−SCs, CMPs, GMPs, and MEPs (Figures 2G–2J, S2F, and S2G). Each stem/progenitor population regenerated from transplanted Lin−CD34+CD38−CD90+CD45RA− cells revealed the same molecular signatures as the same populations isolated directly from the patients (Figure 2K; Figure S2H), establishing the ability of multipotent Lin−CD34+CD38−D90+CD45RA− candidate MDS-SCs to replenish lineage-restricted MDS progenitors.
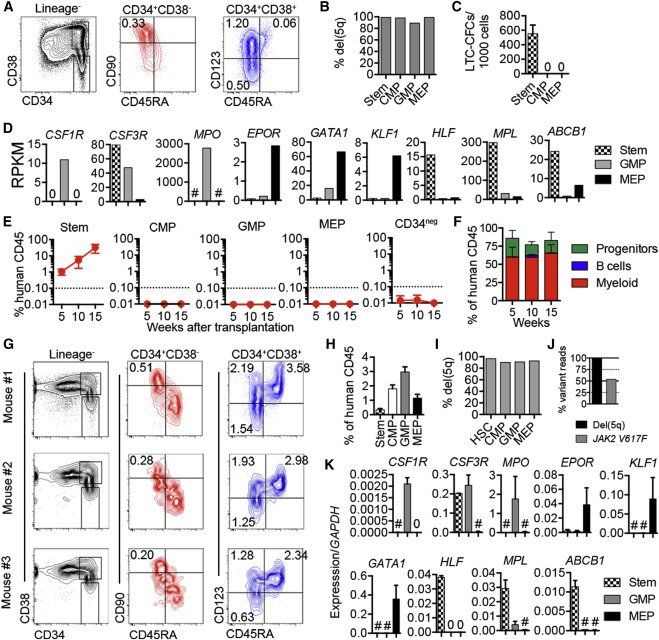
Figure 2.
Hierarchically Organized Stem and Progenitor Cells in del(5q) MDS
(A) FACS profiles and percentages of stem and progenitor cells in total BM from patient 37.
(B) Percentage del(5q) by FISH in purified stem and progenitor cells.
(C) Mean (SEM) LTC-CFC formation from purified stem/progenitor cells from patient 37. Three replicates per population.
(D) RNA sequencing analysis (reads per kilobase per million mapped reads, RPKM) of myeloid, erythroid, and stem cell transcripts in stem/progenitor cells.
(E) Mean (SEM) human (CD45+) engraftment in BM of NSG mice (two to three mice/cell population) transplanted with purified stem/progenitor cells from patient 37.
(F) Mean (SEM) distribution of myeloid (CD15+/CD33+/CD66b+), B cell (CD19+), and CD34+ stem/progenitor cells in MDS SC-derived hCD45 cells (n = two to three mice/donor).
(G) Stem and progenitor profiles in NSG BM of three mice 17–26 weeks after transplantation of purified SCs from patient 37. Percentages within total CD45+ cells are shown.
(H) Mean percentages (SEM) of stem and progenitor cell populations within human CD45+ engrafted cells after transplantation of purified MDS-SCs into NSG mice.
(I) Frequencies of del(5q) by FISH in MDS stem/progenitor cells purified from BM of NSG mice transplanted with purified MDS-SCs.
(J) Variant read frequencies for del(5q) and JAK2V617F in FACS sorted human myeloid (CD15+/CD33+/CD66b+) cells engrafted in NSG mouse 17 weeks after transplantation of purified MDS Lin−CD34+CD38−CD90+CD45RA− SCs.
(K) Mean (SEM) gene expression in stem/progenitor cells purified from BM of NSG mice transplanted with purified MDS-SCs (n = two to three for each population).
See also Figure S2.
Diverse Genetic Lesions in Low- to Intermediate-Risk MDS Originate Exclusively in Rare Lin−CD34+CD38−CD90+CD45RA− MDS-SCs
Recent studies have highlighted the inability of established CSC assays to reliably identify tumor-propagating cells in patients (Clevers, 2011, Kelly et al., 2007, Magee et al., 2012, Quintana et al., 2008 and Taussig et al., 2008), including in MDS (Agarwal, 2012 and ASH-Workshop, 2010). Therefore, to validate MDS-SC identity and activity within patients, we tracked the origin of candidate driver mutations identified in bulk MDS BM cells. Considering the short lifespan of normal myeloid progenitors (Orkin and Zon, 2008), we argued that any stable mutations, contributing to the MDS clone would have been acquired in cells with self-renewal ability. From this it follows that if Lin−CD34+CD38−CD90+CD45RA− cells are the only SCs in low- to intermediate-risk MDS, then the origin of every somatic mutation identified should be traced to this compartment (Figure 3A). If however downstream MDS progenitors have acquired self-renewal ability upon acquisition of driver mutations, we should identify mutations mapped to progenitors but not the upstream Lin−CD34+CD38−CD90+CD45RA− SC compartment (Figures 3B and 3C). Thus, we performed targeted screening for somatic DNA mutations in 83 genes frequently mutated in MDS and other myeloid malignancies (Bejar et al., 2011, Papaemmanuil et al., 2011, Papaemmanuil et al., 2013 and Yoshida et al., 2011).
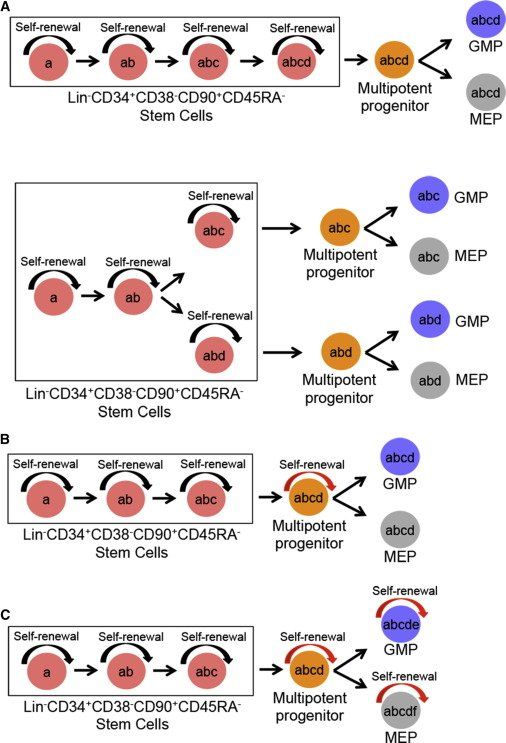
Figure 3.
Alternative Models for Sequential Acquisition of Genetic Lesions in Hierarchically Organized MDS Stem and Progenitor Cells
(A) Only Lin−CD34+CD38−CD90+CD45RA− SCs have self-renewal (inherent; black arrow) ability; and thus all somatic genetic lesions (a–d) can be traced back to the rare SCs regardless whether acquired in a linear (top) or branching (bottom) pattern. As MDS-SCs replenish downstream progenitors, mutations acquired in MDS-SCs are also inherited by their downstream progenitors but without conferring self-renewal potential.
(B) Self-renewal potential has been conferred (red arrow) to a multipotent progenitor cell, which therefore can acquire new and stable genetic lesions (d) not found in the upstream SC compartment but are found in downstream GMPs and MEPs.
(C) Self-renewal potential has been acquired by GMPs and MEPs in addition to the multipotent progenitors thus allowing the occurrence of new stable mutations (e) and (f) in GMPs and MEPs, respectively.
We identified 34 lesions, including del(5q) and mutations in candidate driver-genes in bulk BM cells from 15 patients with low- to intermediate-risk MDS (Figures 4A and 4B; Table S2), including those encoding transcription factors (RUNX1, ETV6), components of signaling pathways (JAK2, CSF3R), epigenetic regulators (TET2, ASXL1), apoptosis regulators (TP53), and spliceosome components (SF3B1, SRSF2,U2AF2, SRSF6). Importantly, these mutations were typically present in the dominant MDS clone as evidenced by a mean variant allele frequency (VAF) in whole BM cells of 30.8% (±3.3%; Figure 4B; Table S2), demonstrating that they must indeed have originated in cells actively propagating the MDS clone in vivo, fulfilling the strictest definition of CSCs ( Clevers, 2011 and Magee et al., 2012). All these genomic lesions were tracked back to the rare Lin−CD34+CD38−CD90+CD45RA− SC compartment, including Lin−CD34+CD38−CD90+CD45RA−-derived long-term colonies ( Figure 4B and S3) and NSG-reconstituting Lin−CD34+CD38−CD90+CD45RA− cells ( Figure 2J). The mean VAF of these somatic mutations was as high as 43.4% (±4.2%) in the Lin−CD34+CD38−CD90+CD45RA− SC compartment, and in all patients, the majority of individual long-term colonies contained all identified genetic driver-lesions, including del(5q) ( Figure 4B). In agreement with this, based on VAF for SNPs in the 5q common deleted region (CDR), a mean of 97.3% (±1.7%) of Lin−CD34+CD38−CD90+CD45RA− SCs in these del(5q) patients were del(5q) ( Figure 4B; Table S2; Supplemental Experimental Procedures). Thus, the clonal advantage of del(5q) MDS over normal hematopoiesis occurs predominantly at the SC-level. As expected based on their hierarchical relationship to Lin−CD34+CD38−D90+CD45RA− MDS-SCs, identified mutations were also found in purified GMPs and MEPs ( Figure 4C). Finally, even if not identified in the bulk BM cells, we screened the purified Lin-CD34+CD38−CD90+CD45RA− SC compartment for reported recurrent driver mutations in MDS ( Bejar et al., 2011, Papaemmanuil et al., 2011 and Yoshida et al., 2011), but never identified a mutation in the Lin−CD34+CD38−CD90+CD45RA− MDS-SC compartment, which was not also represented in bulk MDS cells (P.S.W. and S.E.W.J., unpublished observations).

Figure 4.
Mapping of Somatic Genetic Lesions to Rare and Distinct MDS Stem Cells
(A) del(5q) and genes with mutations in unfractionated (bulk) BM of specified MDS patients identified by targeted sequencing of recurrently mutated genes.
(B) Tracking of mutations identified in whole BM in (A) to Lin−CD34+CD38−CD90+CD45RA− MDS-SCs (stem) and individual SC-derived (LTC-CFC) clones; y-axis numbers indicate percent variant reads for identified mutations. For most patients typical LTC-CFCs produced from CD34+CD38−CD90+CD45RA− MDS-SCs are also shown, harboring all identified driver lesions as indicated by +.
(C) Percentage variant reads for identified mutations in purified GMPs and MEPs from the indicated patients.
See also Figure S3 and Table S2.
del(5q) Precedes Recurrent Driver Mutations in Isolated del(5q) MDS
del(5q) is one of the most frequent cytogenetic aberrations in MDS (Haase, 2008), observed in all subgroups of MDS including, but not restricted to, the distinct subgroup of MDS with isolated del(5q) (previously termed 5q− syndrome; Nimer, 2008). While recent targeted sequencing studies of MDS patients (Bejar et al., 2011,Fernandez-Mercado et al., 2013 and Papaemmanuil et al., 2013) and our data (Figure 4A) suggest that isolated del(5q) MDS cases frequently harbor additional driver mutations, the order of the mutation acquisition relative to del(5q) has not been examined in detail.
To explore this further, we utilized three independent data sets from low- to intermediate-risk MDS cases with del(5q): (1) in-house targeted resequencing of 17 new cases; (2) computational analysis of 36 cases from a previous study (Papaemmanuil et al., 2013), including assessment of the frequencies of cells with del(5q); and (3) nine cases with whole exome sequencing data (Tables S1 and S3). In 19 (36%) of the 53 targeted resequencing cases, no further recurrent driver mutations were identified, a pattern confirmed in five of the nine whole exome-sequenced cases (Figure 5A; Table S3). In 12 of the additional 16 cases with one or more recurrent driver mutations and in which computational analysis allowed high confidence prediction (18 cases did not; Figure S4A), del(5q) was predicted to have occurred as the first genetic lesion, whereas in only four cases was a recurrent driver mutation predicted to have preceded del(5q) (Figure 5B). Notably, in all high-confidence cases where the diagnosis was isolated del(5q) (n = 18) or RAEB-1/RCMD (n = 10), del(5q) was predicted to be the first (or only) genetic lesion, regardless of the identity or number of recurrent driver mutations identified (Figure 5C).
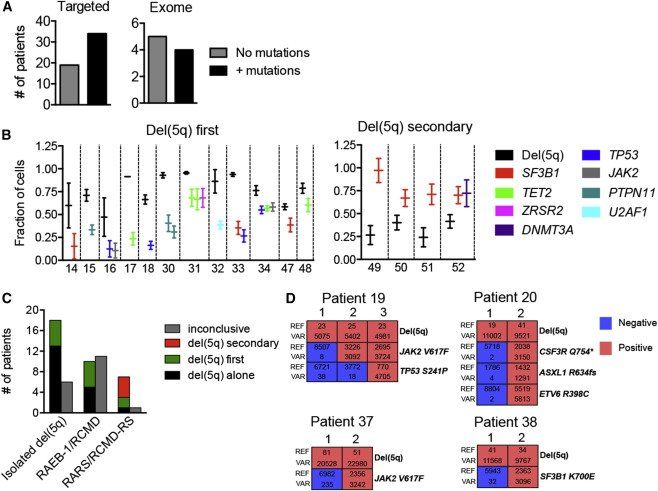
Figure 5.
With Exception of SF3B1-Mutated Cases with Ring Sideroblasts, del(5q) Precedes Recurrent Driver Mutations in Low- to Intermediate-Risk MDS
(A) Number of low- to intermediate-risk del(5q) MDS cases analyzed by targeted (left) or exome (right) sequencing in which no or additional recurrent driver mutations were identified.
(B) High confidence computational prediction of whether or not del(5q) occurred as the first identifiable genetic lesion in low- to intermediate-risk del(5q) MDS cases with one or more candidate driver mutation(s). Error bars represent 95% CI.
(C) Summary of frequencies of del(5q) MDS patient subcategories in which del(5q) was identified as the only, or predicted to be the first or a secondary genetic lesion. Inconclusive, overlap in 95% CI.
(D) Representative single cell (colony) analysis of reference (REF) and variant (VAR) reads for del(5q) and identified driver mutations in four patients with overlapping 95% CI. Red indicates presence and blue absence of del(5q) and the specified mutations in individual colonies (each colony represented by vertical columns).
See also Figure S4 and Table S3.
Eighteen cases showed overlapping estimates of clonal fractions for both del(5q) and the acquired mutations (Figure S4A). To delineate these relationships, we performed single cell analysis in four of these cases, two isolated del(5q) and two RCMD/RAEB-1, and were able to provide evidence that del(5q) also preceded the identified driver mutations in each of these cases (Figure 5D). In contrast, in all four cases in which del(5q) was predicted not to be the initiating genetic lesion, del(5q) was preceded by a recurrent mutation in SF3B1, and all of these patients had been diagnosed with RARS or RCMD-RS ( Figures 5B and 5C).
Recently, compelling evidence for the existence of TIM3− pre-AML SCs was reported, defined as HSCs harboring some but not all recurrent driver mutations identified in the bulk AML, and contributing to balanced lympho-myeloid reconstitution in NSG mice (Jan et al., 2012). Here, we observed no long-term B lymphoid reconstitution in NSG mice transplanted with CD34+ BM cells from nine MDS cases with del(5q) (Figures S4B and S4C), similar to a recent study which also found exclusive long-term myeloid and no B cell contribution from HSCs purified from low-risk MDS patients (Pang et al., 2013). Also, in four del(5q)-MDS patients in which we separated the Lin−CD34+CD38−CD90+ SC compartment into TIM3+ and TIM3− subsets (Figure S4D), we explored their ability to produce myeloid and B cells in vitro. Whereas Lin−CD34+CD38−CD90+ SCs from normal age-matched controls efficiently produced B and myeloid cells, TIM3+ as well as TIM3− SCs from the investigated del(5q) cases produced exclusively myeloid cells (Figure S4E). Thus, in agreement with our findings compatible with del(5q) being the initiating and potentially also the only required genetic lesion for development of isolated del(5q)-MDS, we found no evidence for pre-MDS SCs in low- to intermediate-risk MDS with del(5q).
Evolution of Genetic Lesions in del(5q) MDS Stem Cells during Disease Progression
We next monitored rare Lin−CD34+CD38−CD90+CD45RA− MDS-SCs in sequential BM samples to gain insights into the potential genomic evolution and impact of somatic mutations on the MDS stem and progenitor cell hierarchy, in stable disease as well as prior to disease progression (see Supplemental Experimental Procedures and Table S1). Moreover, to establish a more complete and unbiased picture of somatic exonic mutations, and to what degree diverse mutations (recurrent driver mutations and nonrecurrent, most likely passenger mutations) in the active MDS clones could all be backtracked to the CD34+CD38−CD90+ MDS-SC compartment, we exome-sequenced bulk BM cells of four patients with del(5q) (Figures 6 and 7).
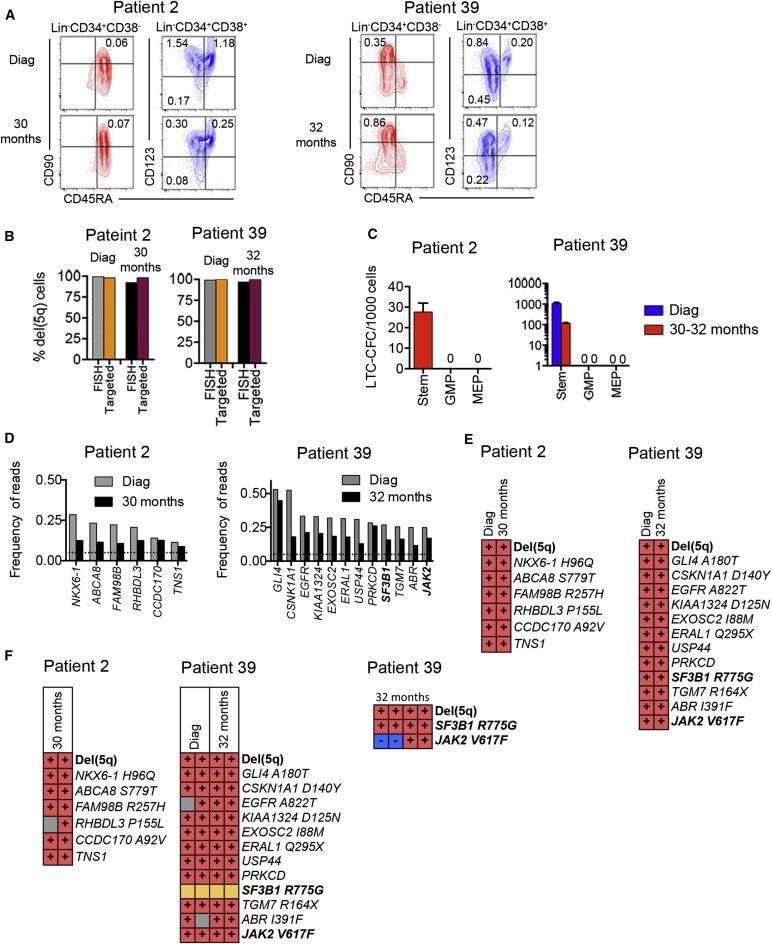
Figure 6.
Mapping of Somatic Mutations to MDS Stem Cells in Sequential Samples in del(5q) MDS without Evidence of Disease Progression
(A) FACS profiles of SCs and progenitors in BM MNCs in serial samples at diagnosis (Diag) and months since diagnosis.
(B) del(5q) clonal involvement of Lin−CD34+CD38−CD90+ MDS-SCs determined by FISH and targeted sequencing.
(C) Mean (SEM) LTC-CFCs generated from Lin−CD34+CD38−CD90+ MDS-SCs, GMPs, and MEPs.
(D) Frequency of exome variant reads in BM MNC. Dotted lines represent 5% variant read cut-off. Recurrent driver mutations are indicated in bold.
(E) Presence (red) and absence (blue) of variant reads for identified mutations in SCs (purified Lin−CD34+CD38−CD90+cells and/or individual LTC-CFCs derived from Lin−CD34+CD38−CD90+ cells). Synonymous mutations are indicated by no specified amino acid change.
(F) del(5q) and mutational status in representative individual Lin−CD34+CD38−CD90+-derived long-term colonies (gray, nonconclusive; orange, primer failure). Colonies shown at the furthest right were analyzed for presence of recurrent driver mutations only. Each vertical column shows mutation status in an individual LTC-CFC.
See also Figure S5 and Tables S4 and S5.
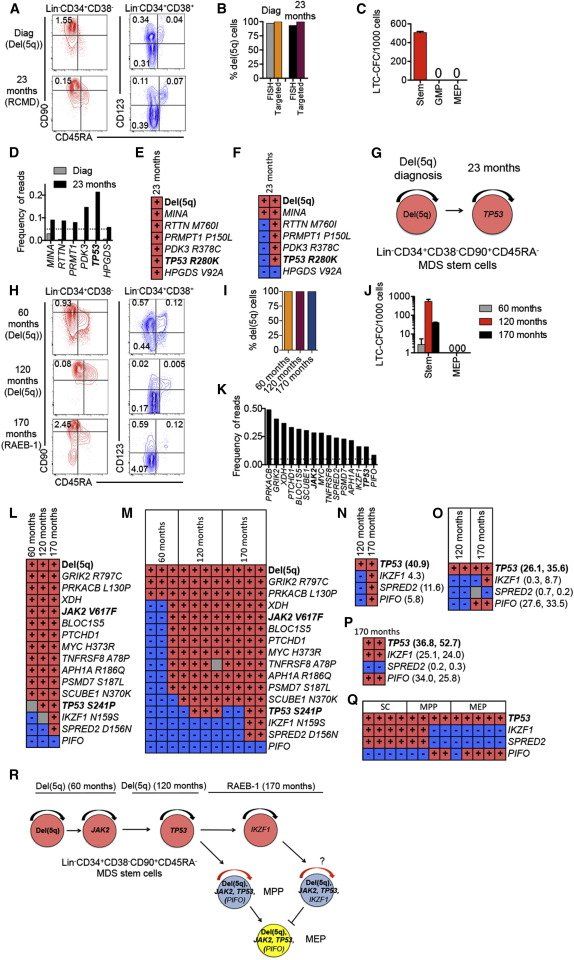
Figure 7.
Genetic Evolution in MDS Stem and Progenitor Cells Preceding AML Transformation
Stem and progenitor cell characterization, and mutational status in sequential BM samples, purified MDS-SCs, and LTC-CFCs from patients 3 (A–G) and 19 (H–R).
(A and H) FACS profiles of SCs and progenitors in BM MNCs in serial samples (at diagnosis and months since diagnosis).
(B and I) del(5q) clonal involvement of Lin−CD34+CD38−CD90+CD45RA− MDS-SCs.
(C and J) Mean (SEM) LTC-CFCs generated from Lin−CD34+CD38−CD90+CD45RA− MDS-SCs, GMPs and MEPs.
(D and K) Frequency of exome variant reads in BM MNC at diagnosis and 23 months (patient 3) and at 170 months (patient 19). Dotted lines represent 5% variant read cut-off. Recurrent driver mutations are indicated in bold.
(E and L) Presence (red) and absence (blue) of variant reads for identified mutations in Lin−CD34+CD38−CD90+ SCs and/or individual LTC-CFCs (gray, nonconclusive).
(F and M) del(5q) and mutational status in representative individual Lin−CD34+CD38−CD90+CD45RA−-derived long-term colonies. Each vertical column shows mutation status in an individual LTC-CFC.
(N–Q) Presence or absence of TP53, IKZF1, SPRED2, and PIFO mutations in BM MNCs (N), MEPs (O), and MPPs (P); two replicate samples analyzed for MEPs and MPPs; percent variant reads at 170 months, are shown to the right, and from representative single cell clones from short-term expanded SCs, MPPs, and MEPs (Q).
(G and R) Proposed sequential acquisition of genetic events based on analysis of sequential samples, individual LTC-CFC, and single-cell SC, MPP, and MEP clones.
See also Supplemental Experimental Procedures, Figure S6, and Tables S6 and S7.
Two of the patients (patients 2 and 39) had stable disease, and exome-sequencing was performed at diagnosis as well as 30–32 months later when they remained transfusion-independent on long-term lenalidomide. At this time, the MDS-SCs, MEPs, and GMPs retained distinct phenotypic, functional, and molecular signatures (Figures 6A–6C; Figure S5). Exome-sequencing identified no (patient 2) and two (patient 39; Figure 6D) recurrent driver mutations, and in both cases a number of predicted somatic passenger mutations (Table S4), which in both patients could all be back-tracked to the Lin−CD34+CD38−CD90+ MDS-SC compartment, including LTC-CFCs, already at diagnosis and none had been eliminated despite the patients being in clinical remission 30–32 months later (Figure 6E and Tables S4 andS5). Sequencing of individual long-term colonies derived from Lin−CD34+CD38−CD90+ SCs established in both patients that all identified mutations had occurred in a linear manner (Figure 6F, Table S5). Notably, neither of these two patients have transformed to AML in the 2–4 years since the last analysis.
In a third, isolated del(5q) case (patient 3) with distinct SC and progenitor signatures (Figures 7A–7C, S6), who transformed to AML only 13 months later (no AML sample available), exome-sequencing of BM cells (23 months after diagnosis) obtained when the patient was responding well to lenalidomide, identified six somatic mutations, including a recurrent TP53 mutation, associated with poor prognosis ( Jädersten et al., 2011). None of these could be confidently identified in bulk BM cells at diagnosis neither by exome (Figure 7D; Table S6) nor targeted ( Table S7) sequencing. However, all identified mutations could be backtracked to the MDS-SC compartment at 23 months ( Figure 7E), and individual LTC-CFC analysis confirmed that del(5q) along with a nonrecurrent MINA mutation preceded the other mutations, including theTP53 mutation ( Figures 7F, 7G, S3, and S6; Table S7).
A fourth patient with isolated del(5q) was analyzed before and following lenalidomide treatment, as well as at disease progression preceding AML transformation (Figures 7H–7Q; Figure S6; Table S1). Exome-sequencing identified 15 somatic mutations at disease progression, including a recurrent JAK2V617F and a recurrent TP53 mutation. The JAK2V617F and ten other nonrecurrent mutations were confidently identified in the bulk BM cells as well as in purified Lin−CD34+CD38−CD90+CD45RA− SCs in the initial BM sample more than 9 years earlier ( Figures 7K and 7L; Tables S6 and S7). Despite reduced transfusion needs on lenalidomide treatment, 5 years later a previously undetectable recurrent TP53 mutation was now detected in bulk BM cells as well as in Lin−CD34+CD38−CD90+CD45RA− SCs ( Figures 7L–7N; Tables S6 and S7). The patient later lost lenalidomide responsiveness and progressed to higher risk MDS (RAEB-1; 9% BM blasts), at which time exome-sequencing identified three additional mutations ( Figures 7K–7N; Tables S6 and S7). Whereas the origin of the new IKZF1 and SPRED2 mutations as other mutations could be mapped to purified Lin−CD34+CD38−CD90+ SCs as well as SC-derived LTC-CFCs, the last mutation (PIFO) detected in 8.5% and 5.8% reads in whole BM with exome and targeted sequencing, respectively ( Figures 7K and 7N; Tables S6 and S7), was undetectable in purified Lin−CD34+CD38−CD90+ SCs and SC-derived LTC-CFCs ( Figures 7L–7M; Table S7). This suggested that the PIFO mutation might have occurred in a progenitor population outside the Lin−CD34+CD38−CD90+ SC compartment, which was likely to have acquired self-renewal potential. Because MEPs and a Lin−CD34+CD38−CD90− multipotent progenitor (MPP) population ( Majeti et al., 2007) had expanded at the time of progression ( Figure 7H), we also performed targeted resequencing of these progenitor populations ( Figures 7O and 7P), and identified, at progression but not in the preceding sample, PIFO variant reads at similar high frequencies as for the TP53 mutation ( Figures 7O and 7P; Table S7). These findings were compatible with the preceding recurrent TP53 mutation that occurred in the Lin−CD34+CD38−CD90+ MDS-SC-compartment, conferring progenitor self-renewal and expansion at disease progression, as signified by the high reads for the predicted silent PIFO mutation exclusively outside the Lin−CD34+CD38−CD90+ compartment, thus here acting as a molecular marker for acquired self-renewal potential outside the Lin−CD34+CD38−CD90+ SC compartment. In further support of this, single cell analysis confirmed the mutually exclusive relationship of the IKZF1 (and SPRED2) and PIFO mutations, representing distinct del(5q) subclones, which prior to this branching had sequentially acquired recurrent del(5q), JAK2 V617F, and TP53 genetic lesions ( Figures 7Q and 7R; Table S7). Notably, this patient transformed to AML 7 months later (from which time no BM or blood sample was available).
DISCUSSION
Efforts to identify distinct human CSCs have become a major focus in translational and clinical cancer research. Consequently, it was a considerable setback, yet to be resolved, when many studies established the inability of in vivo CSC assays to reliably uncover the tumorigenic potential of many cancer cell populations, including in hematological malignancies (Clevers, 2011, Kelly et al., 2007, le Viseur et al., 2008,Magee et al., 2012, Pearce et al., 2006, Quintana et al., 2008 and Taussig et al., 2008). Moreover, the identification of distinct human CSCs exerting their potential in patients (Clevers, 2011 and Magee et al., 2012), including in MDS (Agarwal, 2012 and ASH-Workshop, 2010), has remained elusive.
Here, we provide evidence of rare Lin−CD34+CD38−CD90+ cells functioning as MDS-SCs in patients with low- to intermediate-risk MDS in vivo. Lin−CD34+CD38−CD90+ MDS-SCs are molecularly and functionally distinct from clonally involved GMPs and MEPs. Importantly, Lin−CD34+CD38−CD90+ cells replenish GMPs and MEPs, establishing their hierarchical relationship. Although it cannot formally be ruled out, this hierarchical relationship argues against the possibility of driver mutations occurring outside the Lin−CD34+CD38−CD90+ SC compartment inducing self-renewal potential in progenitors and consequently a Lin−CD34+CD38−CD90+ SC-immunophenotype. Together with these findings, our backtracking of all identified somatic genetic lesions in the bulk BM from patients with MDS to Lin−CD34+CD38−CD90+ MDS-SCs provides evidence for the existence of rare human CSCs in vivo, and that these are the only MDS-SCs in vivo in low-risk MDS. This finding has wide clinical implications, highlighting that propagation and evolution of genetic lesions in the MDS clone is strictly dependent on a rare SC at early stages of MDS. In all investigated cases, the candidate driver mutations were all part of the same dominating SC clone, suggesting that they provide MDS-SCs with a competitive advantage over normal HSCs and the parental MDS clone, but even in combination fail to confer self-renewal potential to downstream progenitors. Even at diagnosis, MDS-SCs typically harbored multiple somatic mutations, including recurrent driver mutations, acquired in a linear, rather than branching, manner within the Lin−CD34+CD38−CD90+ SC compartment.
Sequencing analysis provided insights into the relationship between del(5q) and somatically acquired mutations, in particular in isolated del(5q) MDS. In more than 50% of isolated del(5q) cases, no recurrent driver mutation was identified by targeted or exome sequencing. Although our sequencing strategies may have missed recently identified recurrent mutations in MDS (Klampfl et al., 2013, Nangalia et al., 2013 and Papaemmanuil et al., 2013) and additional significant mutations are likely to be identified in the future, this is unlikely to explain why the fraction of cases without identified recurrent driver mutations is higher in patients with isolated del(5q) than in other groups with low- to intermediate-risk MDS and del(5q). In further support of del(5q) being the initiating genomic lesion in isolated del(5q) MDS, in all cases with at least one identifiable recurrent driver mutation where high confidence sequence of event prediction could be made, del(5q) preceded the acquired mutations. These findings, combined with our lack of evidence for a pre-MDS SC population, and the absence of deletions involving the 5q CDR in a large-scale screen for frequent copy number variations in the blood from older normal individuals (Jacobs et al., 2012), are compatible with del(5q) being an initiating and potentially also the only genomic lesion required to develop the distinct clinical entity of isolated del(5q) MDS. However, further genomic analysis of patients and genetic modeling will be required to unequivocally establish if del(5q) is sufficient to induce isolated del(5q) MDS. Recent studies have shed some light on candidate genes within the 5q CDR responsible for causing some of the phenotypes associated with isolated del(5q) MDS (reviewed in Komrokji et al., 2013). It will be important to explore to what degree haploinsufficiency of the same or distinct genes in the 5q CDR is responsible for the competitive advantage of del(5q) over normal HSCs.
The only low- to intermediate-risk cases in which del(5q) was not the first identified genomic lesion were four cases of ring sideroblastic anemia in which del(5q) was preceded by a recurrent SF3B1 mutation. Intriguingly, just as del(5q) might largely define the phenotype of isolated del(5q) MDS, SF3B1 mutations are thought to be an early and potentially initiating event defining ring sideroblastic anemia ( Papaemmanuil et al., 2011 and Yoshida et al., 2011).
Although limited to four MDS cases with del(5q), tracking CD34+CD38−CD90+MDS-SCs and progenitors by sequential genomic and functional analysis provided insights into the landscape of somatic mutations in stable disease and preceding disease progression, and how they might impact on the MDS stem and progenitor cell hierarchy. In two patients with del(5q) who remained transfusion-independent on long-term lenalidomide treatment, the majority of Lin−CD34+CD38−CD90+ SCs remained part of the del(5q) clone, and no mutations present at diagnosis had been eliminated, nor had any new been acquired. This suggests that in patients with low-risk del(5q) with stable disease, the landscape of somatic mutations in MDS-SCs may be quite stable. However, in two other patients with low-risk isolated del(5q) MDS who later transformed to AML, additional mutations were identified in the SC-compartment while BM blasts remained <5%. In both cases, a recurrent TP53 mutation emerged, conferring worse prognosis ( Jädersten et al., 2011). When one of these patients progressed to RAEB-1 with a higher blast count, additional mutations were mapped to the MDS-SC compartment. However, a predicted passenger mutation (PIFO) could not be mapped to the SC compartment but was instead observed at a high frequency in expanded progenitor compartments along with the precedingTP53 mutation, compatible with acquisition of self-renewal potential by the MPPs (or a progenitor positioned between the Lin−CD34+CD38−CD90+CD45RA− and MPP compartment) at the time of disease progression following acquisition of a TP53 mutation. Although limited to one patient, and awaiting further validation and genetic modeling, this finding provides compelling in vivo evidence in support of sequentially acquired driver genomic lesions (del(5q), JAK2V617F, and TP53) in a small human CSC compartment, eventually conferring self-renewal and thus expansion-potential to downstream progenitors. This is of considerable relevance to the observed disease progression and subsequent AML transformation frequently seen in MDS patients, and in agreement with previous studies implicating that AML frequently contains candidate LSCs with a progenitor rather than SC phenotype ( Goardon et al., 2011 and Jamieson et al., 2004).
From the outset it could not be excluded that progenitors positioned between the Lin−CD34+CD38−CD90+SCs and myeloid-restricted MEPs and GMPs, including MPPs, might also act as MDS-SCs in vivo. However, backtracking of all identified somatic mutations to the Lin-CD34+CD38-CD90+ SC compartment in patients with low-risk MDS suggests that this is rarely the case in early MDS. Our conclusions are limited by the number of patients analyzed, and the fact that patients with MDS are highly heterogeneous with regard to their number and type of recurrent driver mutations (Papaemmanuil et al., 2013). Recent studies in mice have implicated the existence of lineage-biased and perhaps even platelet-myeloid-restricted HSCs (Sanjuan-Pla et al., 2013 and Yamamoto et al., 2013). When corresponding human HSC subsets can be identified, it will be of considerable interest to determine whether mutational targeting of such myeloid-biased or restricted stem cells may explain the largely myeloid-restricted phenotype of MDS.
Our findings suggest that MDS therapies with curative ambitions may best be applied in early MDS when MDS-SCs remain restricted to rare Lin−CD34+CD38−CD90+ cells. Although possessing properties predicting that they will be challenging to target effectively (Tehranchi et al., 2010), the identification of distinct and rare MDS-SCs will facilitate identification of molecular targets and development of targeted therapies to eliminate MDS-SCs in a disease currently notoriously difficult to cure.
Although CSCs in more aggressive human cancers may not be rare, the distinct functional and molecular identity of MDS-SCs established here highlights the critical importance of continued efforts to identify in vivo, monitor and therapeutically target distinct CSCs, as their elimination should not only be essential but, in principle, also sufficient for development of curative cancer therapies.
EXPERIMENTAL PROCEDURES
Patients
Patients with low- to intermediate-risk MDS were included (see Table S1 for clinical details). BM from age-matched healthy individuals was always used for controls unless otherwise specified. All patients provided written informed consent and the study was approved by the ethics committees at the Karolinska Institute, The Norwegian Radiumhospital, Aarhus University Hospital, University of Pavia, Hôpital Avicenne Assistance Publique-Hôpitaux de Paris (AP-HP), University of Dundee, St. James Hospital, University of Oxford, University of New South Wales and Skåne University Hospital.
Flow Cytometry
Stem/progenitor analysis/isolation in BM mononuclear cells was performed as described (Tehranchi et al., 2010) and outlined in the Supplemental Experimental Procedures.
Gene Expression Analysis
Gene expression was analyzed by Fluidigm Dynamic Arrays and global RNA sequencing (Supplemental Experimental Procedures).
FISH
Interphase FISH for del(5q) and +8 was performed as described in the Supplemental Experimental Procedures.
In Vitro Assays
Detailed methods for colony-forming cell (CFC), long-term-culture-CFC (LTC-CFC), and B cell potential, as well as short-term expansion of single cells are described in the Supplemental Experimental Procedures.
Xenograft Transplantation
Cells were transplanted into irradiated NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice, and analyzed as outlined in the Supplemental Experimental Procedures. Experiments were performed in accordance with UK Home Office Project License 30/2570.
DNA Mutational Analysis
Targeted sequencing and exome sequencing for detection, tracking and quantification of genetic lesions were performed and analyzed as described in the Supplemental Experimental Procedures. Briefly, mutations were identified in bulk MDS BM by targeted sequencing of coding region of genes recurrently mutated in myeloid malignancies using a HaloPlex kit (Agilent Technologies) or custom designed RNA baits (Papaemmanuil et al., 2013), as well as by whole exome sequencing using a SureSelect kit (Agilent Technologies) according to the manufacturer’s instructions. Mutations were back-tracked to stem and progenitor compartments by targeted sequencing, either using HaloPlex kit or Access Array chip (Fluidigm) with custom designed PCR primers.
Statistical Analysis
Statistical analysis was performed using Graphpad Prism 6 software. For individual comparisons nonparametric Mann-Whitney test was used and p values less than 0.05 were considered significant. Details of the bioinformatic analysis are included in the Supplemental Experimental Procedures.
AUTHOR CONTRIBUTIONS
S.E.W.J. and P.S.W., with input from S.L., E.P., E.H.-L., and R.S., designed and conceptualized the overall research, analyzed the data, and wrote the manuscript. U.K., O.C., and H.D. contributed equally to these studies and performed and analyzed experiments. D.W., H.D., E.P., S.T., S.L., U.K., R.E., and P.J.C. analyzed and interpreted DNA sequencing data; M.N., R.S., E.G., S.T., Q.D., and I.M. analyzed RNA sequencing data; and K.A., G.G., and B.S. performed FISH analysis. L.S., A.G., and S.D. performed experiments. A.J.M., M.K., C.S., T.M.B., S.N.C., and C.N. advised on experiments. S.-A.C. performed FACS sorting. I.D., D.J., P.F., P.H., M.S.H., M.C., L.M, S.T., D.B., J.B., A.P., J.E.P., A.U., P.V., M.T., G.K., L.N., and E.H.-L. provided patient samples and clinical data. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported by CDP Fellowship from the Leukemia and Lymphoma Society (to P.S.W.), a Wellcome Trust Clinical Fellowship (to O.C.), a Leukemia and Lymphoma Research (LLR) project grant (to S.E.W.J), and a grant from Knut and Alice Wallenberg Foundation to Wallenberg Institute for Regenerative Medicine (to S.E.W.J.). H.D. acknowledges support from a Oxfordshire Healthcare Services Research Training Fellowship and a Wellcome Trust Clinical Research Training Fellowship. J.B. and A.P. acknowledge support from LLR and S.L. acknowledges support from the Swedish Research Council (STARGET) and European Research Council (BRAINCELL, 261063). P.V. is supported by a MRC Disease Team Award and the NIHR Oxford BRC. The authors thank the BMS at Oxford University and the Tayside Tissue Bank, Bishan Wu, Tiphaine Bouriez-Jones, Lillian Wittman, Gunilla Walldin, Monika Jansson, Susan Bray,
Noreen Keenan, Ann Hyslop, Michael Groves Adam Burns, Joanne Mason, Tim Rostron, and Anna Schuh for technical assistance.
ACCESSION NUMBERS
The Gene Expression Omnibus accession number for the RNA sequencing reported in this paper isGSE55689 and the sequence read archive accession number for the DNA sequencing reported in this paper is SRP039353.
Share this Article with others




